Research Themes
The Yang Research Group develops predictive computational frameworks for understanding and designing functional materials. Our work connects atomic-scale physics, electronic structure, symmetry, and chemical complexity to emergent phenomena and materials functionalities.
1. Emergent Phenomena
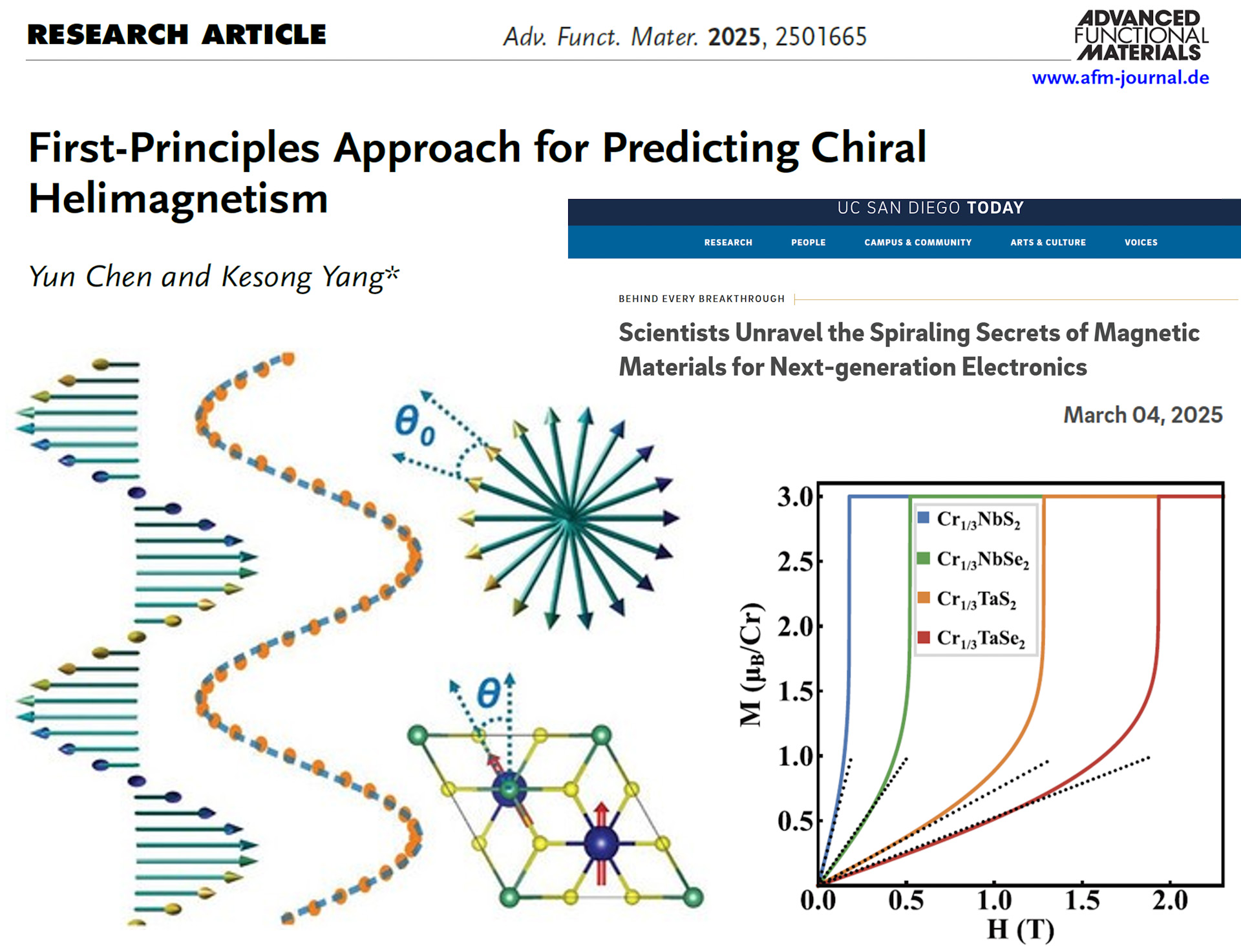
We investigate emergent phenomena in functional materials, including chiral spin textures, collective ordering, interfacial magnetism, and novel magnetic phases. Our research aims to uncover how atomic-scale interactions, symmetry, and electronic structure give rise to complex magnetic and functional behavior in materials.
Representative topics include helimagnetic materials, chiral spin textures, interfacial magnetism, and spintronic functionalities.
2. Structure–Property Coupling
We study how atomic structure, symmetry, dimensionality, and interfacial environments govern electronic and optical functionalities in materials. Our work focuses on uncovering structure–property relationships that enable emergent responses in semiconductors, oxide heterostructures, and hybrid functional systems.
Representative directions include electronic structure engineering, symmetry-driven functionalities, and interfacial electronic phenomena.
3. Predictive Materials Discovery
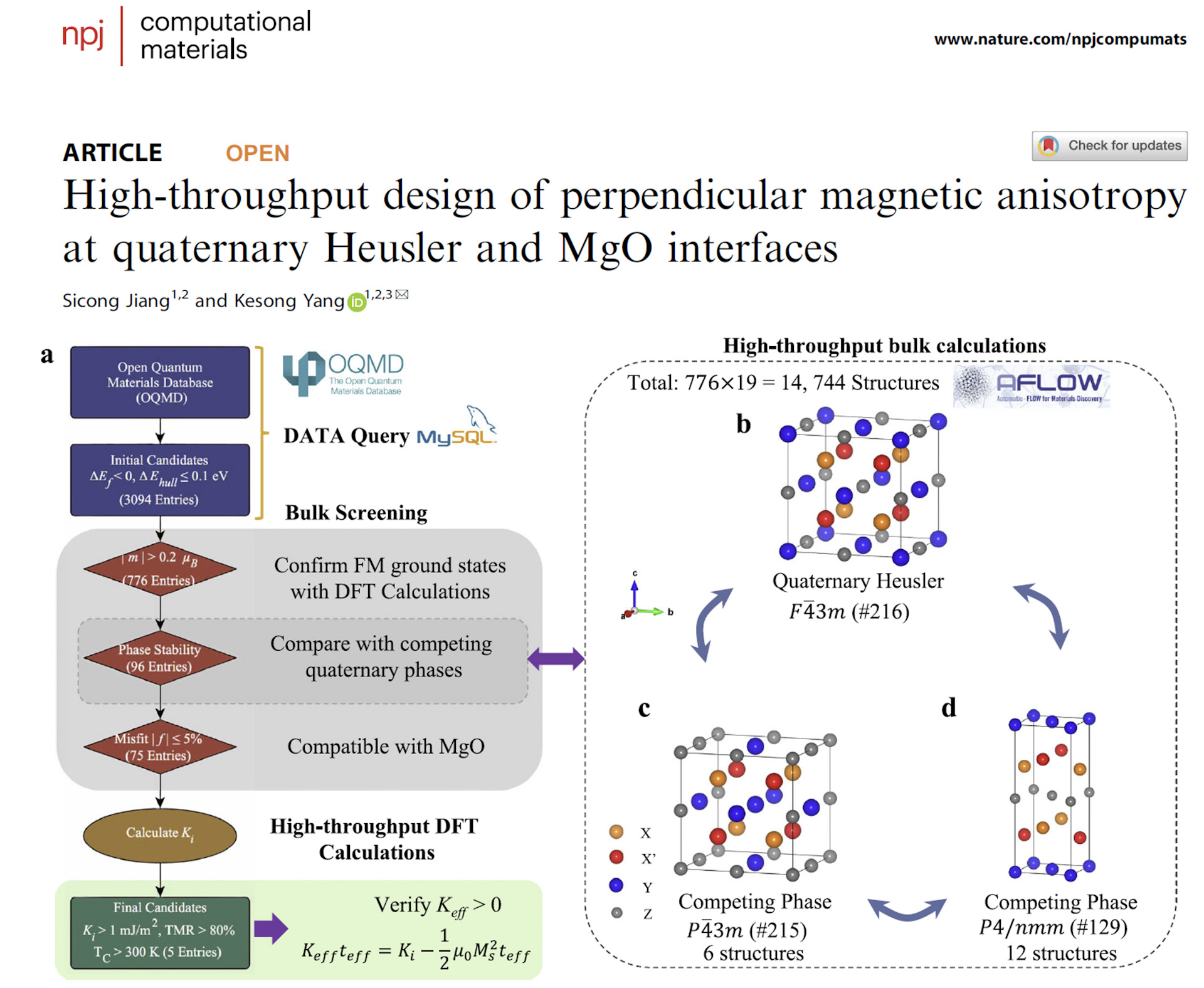
We develop scalable computational frameworks for predictive materials discovery through high-throughput screening, automated workflows, and descriptor-guided exploration. By integrating first-principles calculations with data-driven methodologies and physics-guided modeling, we accelerate the discovery of functional materials across magnetism, electronics, energy, and optical technologies.
Current efforts include high-throughput screening of functional magnets, semiconductors, interfaces, and emergent materials systems.

4. Complex Materials Systems
Many technologically important materials contain disorder, interfaces, defects, and structural complexity across multiple length scales. Our research develops first-principles structure modeling approaches for amorphous, disordered, and interfacial materials, with emphasis on realistic atomic-scale representations of complex materials systems.
Current directions include grain-boundary modeling, amorphous and semi-amorphous materials, interface engineering, and physically realistic structure construction.

Density functional theory, electronic structure, polarization, magnetism, defects, interfaces, and disorder.
Automated screening, AFLOW-related workflows, database-driven discovery, and scalable computational pipelines.